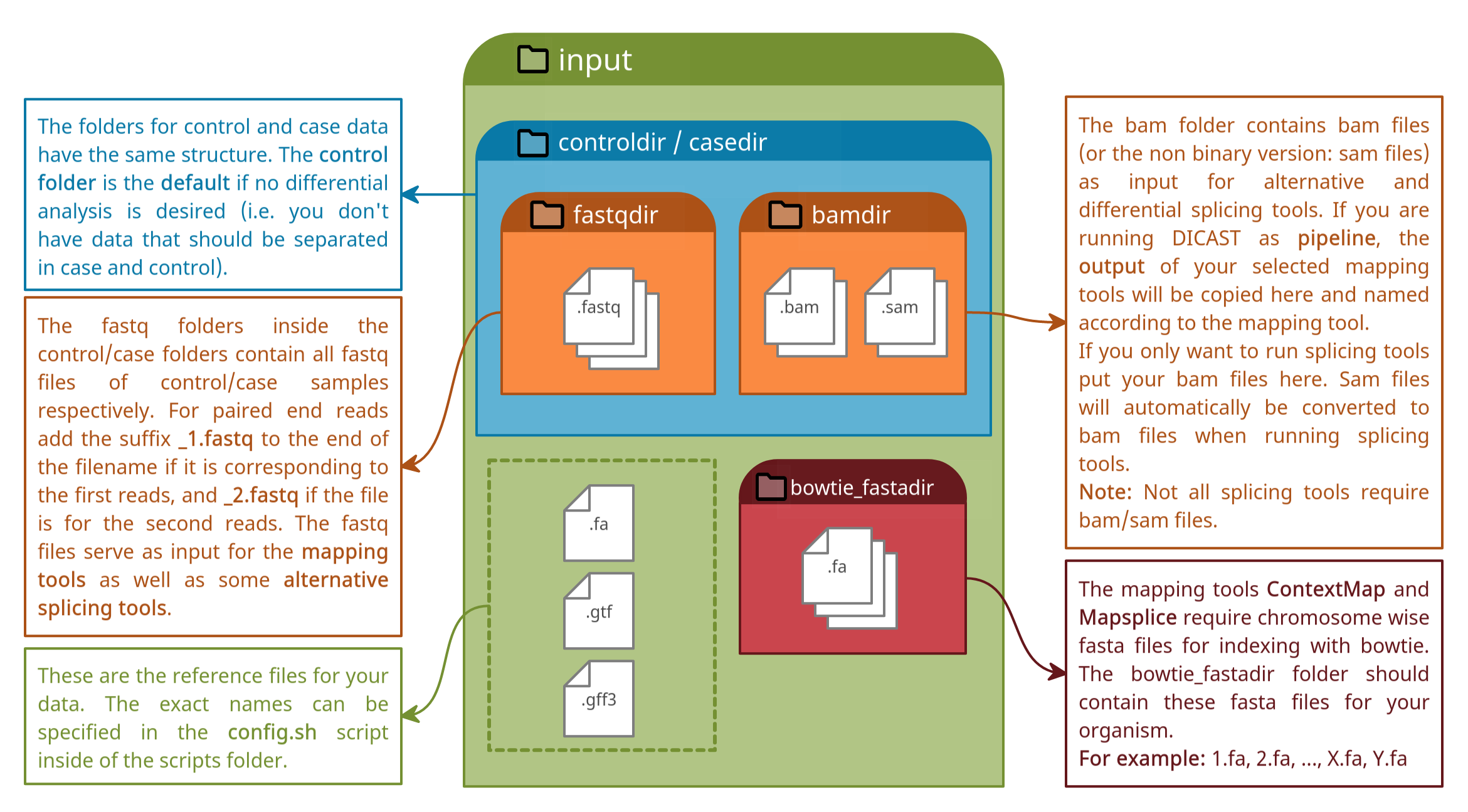
Directory Structure
Note
Our pipeline allows to run many different tools in the same way. The scripts therefore rely on the directory structure specified here. Please don’t rename any directories that are listed here within the git. An output directory is created with your first run. This directory may be renamed.
Example Tree Structure
This is an example for the tree structure when running the pipeline for alternative splicing. Please note that you only need a .fa and a .gtf file if you start your analysis with ASimulator, since it will create .fastq files for you. However, some tools need specific input files. Please refer to the respective tool documentation for further information.
input
├── casedir
│ ├── bamdir
│ └── fastqdir
├── controldir
│ ├── bamdir
│ │ ├── example_bbmap.sam
│ │ ├── example_contextmap.sam
│ │ ├── example_crac.sam
│ │ ├── example_dart.sam
│ │ ├── example_gsnap.sam
│ │ ├── example_hisat.sam
│ │ ├── example_mapsplice.sam
│ │ ├── example_minimap.sam
│ │ ├── example_segemehl.sam
│ │ ├── example_starAligned.out.sam
│ │ └── example_subjunc.sam
│ └── fastqdir
│ ├── example_1.fastq
│ └── example_2.fastq
├── bowtie_fastadir
│ ├── 10.fa
│ ├── 11.fa
│ ├── 12.fa
│ ├── 13.fa
│ ├── 14.fa
│ ├── 15.fa
│ ├── 16.fa
│ ├── 17.fa
│ ├── 18.fa
│ ├── 19.fa
│ ├── 1.fa
│ ├── 20.fa
│ ├── 21.fa
│ ├── 22.fa
│ ├── 2.fa
│ ├── 3.fa
│ ├── 4.fa
│ ├── 5.fa
│ ├── 6.fa
│ ├── 7.fa
│ ├── 8.fa
│ ├── 9.fa
│ ├── MT.fa
│ ├── X.fa
│ └── Y.fa
├── example.fa
├── example.gff
└── example.gtf
Input files
The sample_input is a template of what the files should look like. Let’s however compare this with a real world example. DICAST is not intended to be limited to any specific organism, but for examples, we go with the assembly you can download at NCBI homo sapiens.
example.fa : refers to a reference genome .fna or fa.
example.gff: refers to a reference annotation .gff or .gff3.
example.gtf: refers to a reference annotation .gtf.
Warning
All files, including references and fastq files must be unzipped, as most of the tools within dicast require them in unzipped form.
Note
If you work with the human genome or would like to just test if DICAST is installed well, check out the script at initializing-dicast.sh and execute it with the command bash initializing-dicast.sh, to populate your input directory with relavent human references.
Note
casedir, is currently unsupported. DICAST was built originally with a design that included tools for differential analysis. It maintains the directory structure in order to expand, to cover differential tools in the future.
example_*.sam: DICAST can map your fastq files for you with a mapper of your choice, the results of such mapping will be found here in this directory bamdir. If you have already mapped bam/sam files, place them in the input/controldir/bamdir, for DICAST to start from here.