Run your analysis
Note
This guide page assumes that you have followed all four pages of the setup carefully and that you have configred DICAST.
Workflow
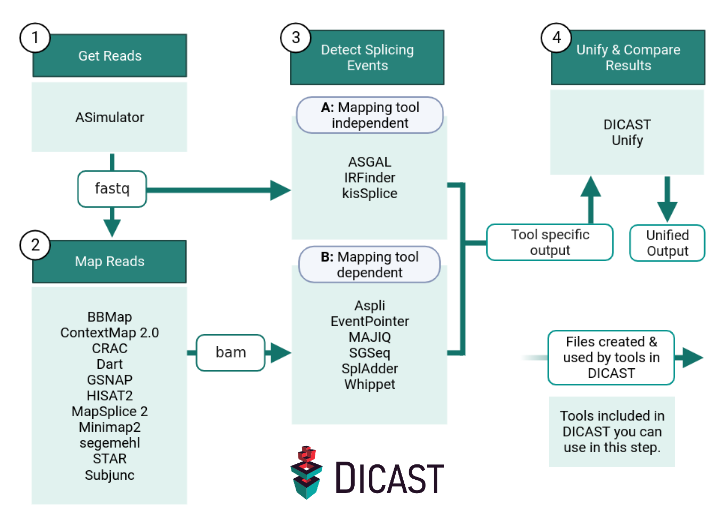
To run the entire pipeline, you need a reference genome file and a annotation file file of your organism. If you do not want to work with simulated data, you can enter each step with your own data. E.g. you can enter step 2 or 3A with fastq files from your own experiment or step 3B with bam files from your own mapping tool. Please note that not all mapping and splicing detection tools are compatible with each other and have different file requirements (e.g. reference genome, annotation file, gff). For further information, please refer to the tool-specific DICAST documentation.
Warning
We tried our best to unify the input that is required for all tools. This did not work for all tools. When a tool requires custom input you will see a warning like this on the concerning documentation page.